Introduction
This notebook will show you how to work with the C. elegans L2-stage sci-RNA-seq data from Cao et al. (Science, 2017). It aims to cover the following use cases:
- Accessing the raw data
- Exploring the expression pattern of a gene interest
- Finding differentially expressed genes between subsets of cells
- Re-clustering subsets of the data using t-SNE
Key advantages of sci-RNA-seq over contemporary alternatives (such as droplet-based single cell RNA-seq):
- sublinear cost scaling
- reliance on widely available reagents and equipment
- ability to concurrently process many samples within a single workflow
- compatibility with methanol fixation of cells
- cell capture based on DNA content rather than cell size
- the flexibility to profile either cells or nuclei
Installation
Required R Packages
- dplyr
- ggplot2
- monocle version 2.3.5
Monocle is a comprehensive package for single cell analysis developed by the Trapnell lab. Monocle version 2.3.5 is the version that was used for the paper. To install Monocle 2.3.5, open a command line and run:
curl -O http://waterston.gs.washington.edu/sci_RNA_seq_gene_count_data/monocle_2.3.5.tar.gz
R CMD INSTALL monocle_2.3.5.tar
Later versions of Monocle may produce different results for use case 4 (re-clustering with t-SNE) due to changes in how we preprocess the data before running t-SNE. We will update this vignette when we release the next version of Monocle, 2.6.0, to support the new version. If you wish to learn more about Monocle click the following link to go to the Monocle Website .
Now, go back to your R session and verify that Monocle 2.3.5 installed successfully (look for monocle_2.3.5 in the output of sessionInfo). suppressPackageStartupMessages({
library(monocle)
library(dplyr)
library(ggplot2)
})
sessionInfo()
Next, we'll download an RData file that has the Cao et al. data and some utility functions to help navigate it.
download.file(
"http://waterston.gs.washington.edu/sci_RNA_seq_gene_count_data/Cao_et_al_2017_vignette.RData",
destfile = "Cao_et_al_2017_vignette.RData")
load("Cao_et_al_2017_vignette.RData")
ls()
Important Information
- cds is a Monocle CellDataSet object containing the single cell RNA-seq data from the main L2-stage C. elegans experiment described in Cao et al., along with annotations.
- cds.neurons is a re-clustered subset of the neuronal cells from cds.
- cds.experiment.2 has data from the second C. elegans experiment described in Cao et al.. This includes intestine cells that were missed in the first experiment, but the data overall is lower quality than the first experiment. In the manuscript, we only included the intestine cells from this experiment and excluded the rest.
Do Not Open GitHub Issues for Questions
If you need help or have any questions, submit your posts to the Google Group. If you find a problem with the code in the vignettes or with the data quality, open a GitHub issue.
Use Case 1: Accessing the Raw Data
To this end, we have created a website to facilitate the further annotation of these data by the community (http://atlas.gs.washington.edu). Gene-by-cell matrices and vignettes for how to work with the data are also hosted at this site.
If you aren't familiar with working with single cell RNA-seq data, we highly recommend that you take a look at the examples and utility functions presented in the other sections of this document instead of trying to dive in to the raw data directly.
exprs(cds)[1:3, 1:3]
| cele-001-001.CATGACTCAA | cele-001-001.AAGACGGCCA | |
|---|---|---|
| WBGene00000001 | . | . |
| WBGene00000002 | . | . |
| WBGene00000003 | . | . |
| cele-001-001.GCCAACGCCA | ||
| WBGene00000001 | . | |
| WBGene00000002 | . | |
| WBGene00000003 | . |
fData(cds)[1:3,]
| gene_id | symbol | num_cells_expressed | |
|---|---|---|---|
| WBGene00000001 | WBGene00000001 | aap-1 | 1016 |
| WBGene00000002 | WBGene00000002 | aat-1 | 354 |
| WBGene00000003 | WBGene00000003 | aat-2 | 897 |
The pData function is used to access cell annotations.
| annotation | description |
|---|---|
n.umi |
the number of unique molecular identifiers observed to be expressed by a given cell |
Size_Factor |
n.umi divided by the geometric mean of n.umi across all cells |
tsne_1 and tsne_2 |
the coordinates for the cell in the t-SNE dimensionality reduction |
Cluster |
the cluster id assigned by the density peak clustering algorithm |
cell.type and tissue |
the cluster id assigned by density peak clustering algorithm applied on the first two t-SNE reduced dimensions |
pData(cds)[1:3,]
| cele-001-001.CATGACTCAA | cele-001-001.AAGACGGCCA | cele-001-001.GCCAACGCCA | |
|---|---|---|---|
| cell | cele-001-001.CATGACTCAA | cele-001-001.AAGACGGCCA | cele-001-001.GCCAACGCCA |
| n.umi | 144 | 790 | 832 |
| plate | 001 | 001 | 001 |
| Size_Factor | 0.2368328 | 1.2992911 | 1.3683674 |
| num_genes_expressed | 89 | 419 | 338 |
| tsne_1 | 5.4866377 | -3.8619751 | -0.5594413 |
| tsne_2 | 14.67085 | -27.63448 | 41.98569 |
| Cluster | 20 | 6 | 13 |
| peaks | FALSE | FALSE | FALSE |
| halo | TRUE | TRUE | TRUE |
| delta | 0.02491657 | 0.40961274 | 0.04445184 |
| rho | 893.9855 | 812.2076 | 240.2908 |
| cell.type | Unclassified neurons | Germline | Intestinal/rectal muscle |
| tissue | Neurons | Gonad | Intestinal/rectal muscle |
neuron.type in pData(cds.neurons) is the annotation used in Figure 4 of Cao et al.
| cele-001-001.CATGACTCAA | cele-001-001.AACTACGGCT | cele-001-001.GAGGCTTATT | |
|---|---|---|---|
| cell | cele-001-001.CATGACTCAA | cele-001-001.AACTACGGCT | cele-001-001.GAGGCTTATT |
| n.umi | 144 | 201 | 117 |
| plate | 001 | 001 | 001 |
| Size_Factor | 0.2368328 | 0.3305791 | 0.1924267 |
| num_genes_expressed | 89 | 129 | 76 |
| tsne_1 | 0.9574604 | -3.0567593 | -18.5689290 |
| tsne_2 | 0.8288424 | -41.4083795 | -33.9833909 |
| Cluster | 11 | 8 | 39 |
| peaks | FALSE | FALSE | FALSE |
| halo | TRUE | TRUE | TRUE |
| delta | 0.37046400 | 0.25861943 | 0.02962754 |
| rho | 108.71265 | 70.88069 | 37.29414 |
| cell.type | Unclassified neurons | Ciliated sensory neurons | Ciliated sensory neurons |
| tissue | Neurons | Neurons | Neurons |
| neuron.type | Cholinergic (11) | ASI/ASJ | AFD |
Use Case 2: Expression Pattern of a Gene of Interest
The show.expr.info function returns statistics related to the expression of a given gene in tabular form.
The first argument is the gene name.
The second argument specifies whether to show statistics at the level of tissues, cell types, or neuron (sub)-types.
See the examples below.
the function returns a data frame with five columns:
| name | description |
|---|---|
facet |
the tissue / cell type / neuron type |
tpm |
the expression of the given gene in the facet in TPM (transcripts per million) |
prop.cells.expr |
the proportion of cells in the facet that express at least one UMI (unique molecular identifier) for the given gene. Note that cells in different facets can have very different average number of UMIs per cell, so TPM is the better measure of relative expression. |
n.umi |
the number of UMIs (unique molecular identifiers) observed for the given gene in the facet. This is the "sample size" from which the TPM value is computed |
total.n.umi.for.facet |
the total number of UMIs observed across all genes for all cells in the facet |
RNA Leakage
Note that low levels of expression of a given gene in cell types you would not expect may be due to leakage of RNA in the fixed cells. For this experiment, based on the expression patterns for a few marker genes, we estimate that the background level of expression you will observe randomly in cells that do not truly express a given gene is ~1-2% of the expression of the highest-expressing cell type.
show.expr.info("emb-9", "tissue")
| facet | tpm | props.cells.expr | n.umi | total.n.umi.for.facet |
|---|---|---|---|---|
| Body wall muscle | 7972.27222 | 0.96451347 | 157028 | 19390434 |
| Gonad | 390.16652 | 0.03757116 | 3597 | 11166871 |
| Intestine | 597.88569 | 0.09775967 | 102 | 1230975 |
| Neurons | 89.83882 | 0.01406926 | 187 | 2203067 |
| Gila | 72.96189 | 0.02148228 | 39 | 787560 |
| Pharynx | 165.1210 | 0.01592357 | 14 | 85381 |
| Hypodermis | 47.05950 | 0.01821904/th> | 158 | 5821384 |
show.expr.info("emb-9", "cell type") %>% head(10)
| facet | tpm | prop.cells.expr | n.umi | total.n.umi.for.facet |
|---|---|---|---|---|
| Distal Tip Cells | 16165.1598 | 0.97520661 | 3405 | 202581 |
| Body wall muscle | 7972.2722 | 0.96451347 | 157028 | 19390434 |
| Intestinal/rectal muscle | 5211.8861 | 0.84740260 | 2622 | 439170 |
| Sex myoblasts | 218.2425 | 0.14776632 | 75 | 377288 |
| Pharyngeal neurons | 165.1210 | 0.01592357 | 14 | 85381 |
| Other interneurons | 137.8986 | 0.02483070 | 17 | 172852 |
| Socket Cells | 123.7517 | 0.02793296 | 26 | 184774 |
| Coelomocytes | 115.4683 | 0.02503682 | 71 | 544263 |
| Non-seam hypodermis | 102.4186 | 0.02006689 | 56 | 1059546 |
| Somatic gonad precursors | 102.1766 | 0.06376812 | 75 | 823856 |
show.expr.info("R102.2", "neuron type") %>% head(10)
| facet | tpm | prop.cells.expr | n.umi | total.n.umi.for.facet |
|---|---|---|---|---|
| Cluster 21 | 9807.74014 | 0.91588785 | 446 | 49587 |
| Cluster 16 | 8266.64832 | 0.66025641 | 363 | 47719 |
| ASK | 6720.61429 | 0.81944444 | 155 | 24157 |
| ASI/ASJ | 6482.22237 | 0.74358974 | 239 | 40396 |
| ASG | 2121.04289 | 0.39534884 | 48 | 26295 |
| ASEL | 1670.50501 | 0.37837838 | 17 | 11042 |
| ASER | 795.91129 | 0.28571429 | 16 | 15324 |
| AWB/AWC | 85.09161 | 0.02380952 | 4 | 23186 |
| Cholinergic (15) | 60.56935 | 0.01538462 | 1 | 14463 |
| Pharyngeal (33) | 58.35581 | 0.02857143 | 2 | 25101 |
Source Code of Utility Functions
Note that you can see the source code for any of the utility functions used in this vignette. Just run the function name with no parentheses.
show.expr.info
function (gene, expr.info)
{
if (class(expr.info) == "character") {
expr.info = gsub("[.]", " ", tolower(expr.info))
if (expr.info == "tissue")
expr.info = tissue.expr.info
else if (expr.info == "cell type")
expr.info = cell.type.expr.info
else if (expr.info == "neuron type")
expr.info = neuron.type.expr.info
}
gene.id =
get.gene.id(gene, fData.df = expr.info$gene.annotations)
data.frame(facet = names(expr.info$tpm[gene.id, ]),
tpm = expr.info$tpm[gene.id,],
prop.cells.expr = expr.info$prop.cells.expr[gene.id,],
n.umi = expr.info$n.umi[gene.id, ],
total.n.umi.for.facet = expr.info$total.n.umi.for.facet)
%>% arrange(-tpm)
}
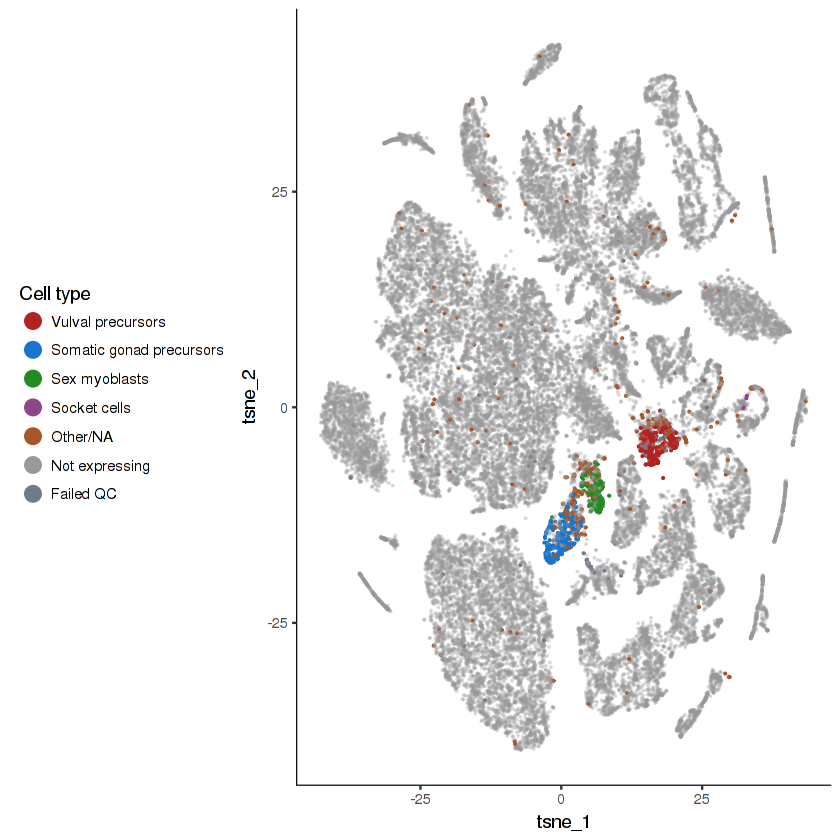
- Cells that do not express the gene will be colored grey. They are made semi-transparent so as to better highlight the cells that do express the gene.
- Cells that express the gene will be colored according to their cell type. The top 4 highest-expressing cell types will be assigned distinct colors. Other cell types will be lumped together.
- Cells in the "Failed QC" category are those that express the gene, but are excluded from the analysis due to either having an unusually low UMI count or being a likely doublet.
plot.expr(cds, "lin-12")
plot.expr(cds.neurons, "che-3")
Use Case 3: Differential expression between cell subsets
We've defined a function two.set.differential.gene.test that finds and reports statistics on differentially expressed genes (DEG) that distinguish between two defined sets of cells. This is a wrapper that just adds a bit of functionality around Monocle's differentialGeneTest function.
two.set.differential.gene.test takes four arguments:| argument | description |
|---|---|
cds |
a CellDataSet object that includes both sets of cells you wish to compare |
set.1.filter |
a boolean vector of length ncol(cds) indicating which cells should be in Set 1 |
set.2.filter |
a boolean vector of length ncol(cds) indicating which cells should be in Set 2 |
formal |
if TRUE, p-values and q-values are computed for differential gene expression and genes are ranked by q-value. if FALSE, no formal statistical test is performed and genes are heuristically ranked. formal = F makes the function run much, much faster |
Large Memory Use for Differential Expression Tests
If you run two.set.differential.gene.test on large sets of cells (> 1000-ish), it may take a bunch of memory. The utility functions is.tissue, is.cell.type, and is.neuron.type may be used to create the boolean vectors required for the set.1.filter and set.2.filter parameters. Each function takes a CellDataSet and a string and tests for each cell whether its tissue / cell type / neuron type is defined (not NA) and equal to the given value.
is.tissue
function (cds, x)
{
with(pData(cds), !is.na(tissue) & tissue == x)
}
head(is.tissue(cds, "Gonad"))
> FALSE TRUE FALSE FALSE FALSE TRUE
sum(is.tissue(cds, "Gonad"))
> 5628
sum(is.cell.type(cds, "Distal tip cells"))
sum(is.neuron.type(cds.neurons, "ASEL"))
> 129
> 37
tissues
> 'Body wall muscle' 'Pharynx' 'Hypodermis' 'Neurons' 'Gila' 'Gonad' 'Intestine'
cell.types
> 'Am/PH sheath cells' 'Body wall muscle' 'Canal associated neurons'
'Cholinergic neurons' 'Ciliated sensory neurons' 'Coelomocytes' 'Distal tip cells'
'Dopaminergic neurons' 'Excretory cells' 'flp-1(+) interneurons'
'GABAergic neurons' 'Germline' 'Intestinal/rectal muscle' 'Intestine'
'Non-seam hypodermis' 'Other interneurons' 'Oxygen sensory neurons'
'Pharyngeal epithelia' 'Pharyngeal gland' 'Pharyngeal muscle' 'Pharyngeal neurons'
'Rectum' 'Seam cells' 'Sex myoblasts' 'Socket cells' 'Somatic gonad precursors'
'Touch receptor neurons' 'Vulval precursors'
neuron.types
'AFD' 'ASEL' 'ASER' 'ASG' 'ASI/ASJ' 'ASK' 'AWA' 'AWB/AWC' 'BAG' 'CAN'
'Cholinergic (11)' 'Cholinergic (15)' 'Cholinergic (23)' 'Cholinergic (24)'
'Cholinergic (26)' 'Cholinergic (29)' 'Cholinergic (3)' 'Cholinergic (35)'
'Cholinergic (36)' 'Cluster 10' 'Cluster 13' 'Cluster 16' 'Cluster 17'
'Cluster 21' 'Cluster 25' 'Cluster 27' 'Cluster 40' 'Cluster 5' 'Dopaminergic'
'DVA' 'flp-1(+)' 'GABAergic' 'Pharyngeal (33)' 'Pharyngeal (37)' 'PVC/PVD'
'RIA' 'RIC' 'SDQ/ALN/PLN' 'Touch receptor' 'URX/AQR/PQR'
ASEL.vs.ASER.DEG = two.set.differential.gene.test(
cds.neurons, is.neuron.type(cds.neurons, "ASEL"),
is.neuron.type(cds.neurons, "ASER"))
two.set.differential.gene.test returns the following statistics:
| argument | description |
|---|---|
set.1.umi |
the number of UMI (unique molecular identifiers) observed for the gene in Set 1 |
set.2.umi |
the number of UMI (unique molecular identifiers) observed for the gene in Set 2 |
set.1.tpm |
gene expression in Set 1 in TPM (transcripts per million) |
set.2.tpm |
set.2.tpm -- gene expression in Set 2 in TPM (transcripts per million) |
higher.expr |
the number of UMI (unique molecular identifiers) observed for the gene in Set 1 |
log2.ratio |
the number of UMI (unique molecular identifiers) observed for the gene in Set 2 |
precision |
gene expression in Set 1 in TPM (transcripts per million) |
recall |
set.2.tpm -- gene expression in Set 2 in TPM (transcripts per million) |
f.score |
harmonic mean of precision and recall. genes are sorted by this metric |
ASEL.vs.ASER.DEG %>% head()
| gene | tank-1 | gcy-22 | gei-3 | gcy-3 | gcy-6 | T27C4.1 |
|---|---|---|---|---|---|---|
| set.1.n.umi | 20 | 0 | 10 | 0 | 66 | 30 |
| set.1.n.umi | 570 | 129 | 166 | 147 | 0 | 164 |
| set.1.tpm | 1794.188 | 0.000 | 1095.928 | 0.000 | 6909.705 | 3210.293 |
| set.2.tpm | 36169.778 | 8862.360 | 12076.301 | 9593.506 | 0.000 | 11338.218 |
| higher.expr | Set 2 | Set 2 | Set 2 | Set 2 | Set 1 | Set 2 |
| log2.ratio | 4.332578 | 13.113475 | 3.460638 | 13.227842 | 12.754408 | 1.819968 |
| precision | 0.8536585 | 1.0000000 | 0.9090909 | 1.0000000 | 1.0000000 | 0.6808511 |
| recall | 1.0000000 | 0.8000000 | 0.8571429 | 0.7428571 | 0.7297297 | 0.9142857 |
| f.score | 0.9210526 | 0.8888889 | 0.8823529 | 0.8524590 | 0.8437500 | 0.7804878 |
ASEL.vs.ASER.DEG %>% filter(higher.expr == "Set 1")
%>% head()
| gene | gcy-6 | gcy-17 | crh-1 | gcy-20 | gcy-7 | unc-44 |
|---|---|---|---|---|---|---|
| set.1.n.umi | 66 | 69 | 58 | 53 | 39 | 88 |
| set.2.n.umi | 0 | 0 | 12 | 0 | 0 | 45 |
| set.1.n.tpm | 6909.705 | 6553.874 | 5675.336 | 5339.037 | 3709.660 | 7647.438 |
| set.2.tpm | 0.000 | 0.000 | 800.504 | 0.000 | 0.000 | 2915.088 |
| higher.expr | Set 1 | Set 1 | Set 1 | Set 1 | Set 1 | Set 1 |
| log2.ratio | 12.754408 | 12.678132 | 2.823924 | 12.382364 | 11.857071 | 1.390942 |
| precision | 1.0000000 | 1.0000000 | 0.8333333 | 1.0000000 | 1.0000000 | 0.6000000 |
| recall | 0.7297297 | 0.6216216 | 0.6756757 | 0.5945946 | 0.5675676 | 0.8108108 |
| f.score | 0.8437500 | 0.7666667 | 0.7462687 | 0.7457627 | 0.7241379 | 0.6896552 |
Running two.set.differential.gene.test with formal = T will compute q-value (the false detection rate at which a gene can be considered to be differentially expressed between the two sets). This is very slow however, so we recommend not using it for exploratory analysis and only using it after you've found something interesting and want to verify that the finding is statistically robust.
system.time({
ASEL.vs.ASER.DEG = two.set.differential.gene.test(
cds.neurons,
is.neuron.type(cds.neurons, "ASEL"),
is.neuron.type(cds.neurons, "ASER"),
formal = T, cores = min(16, detectCores()))
})
| user | system | elapsed |
|---|---|---|
| 21.241 | 3.599 | 152.575 |
ASEL.vs.ASER.DEG %>% head()
| gene | tank-1 | gcy-22 | gei-3 | gcy-3 | gcy-6 | T27C4.1 |
|---|---|---|---|---|---|---|
| set.1.n.umi | 20 | 0 | 10 | 0 | 66 | 30 |
| set.2.n.umi | 570 | 129 | 166 | 147 | 0 | 164 |
| set.1.tpm | 1794.188 | 0.000 | 1095.928 | 0.000 | 6909.705 | 3210.293 |
| set.2.tpm | 36169.778 | 8862.360 | 12076.301 | 9593.506 | 0.000 | 11338.218 |
| higher.expr | Set 2 | Set 2 | Set 2 | Set 2 | Set 1 | Set 2 |
| log2.ratio | 4.332578 | 13.113475 | 3.460638 | 13.227842 | 12.754408 | 1.819968 |
| precision | 0.8536585 | 1.0000000 | 0.9090909 | 1.0000000 | 1.0000000 | 0.6808511 |
| recall | 1.0000000 | 0.8000000 | 0.8571429 | 0.7428571 | 0.7297297 | 0.9142857 |
| f.score | 0.9210526 | 0.8888889 | 0.8823529 | 0.8524590 | 0.8437500 | 0.7804878 |
touch.receptor.neurons.DEG %>% filter(higher.expr == "Set 1") %>% head()
| gene | mec-17 | mec-18 | mtd-1 | mec-7 | mec-1 | mec-9 |
|---|---|---|---|---|---|---|
| set.1.n.umi | 2720 | 796 | 443 | 4418 | 1717 | 726 |
| set.2.n.umi | 118 | 49 | 15 | 743 | 695 | 337 |
| set.1.tpm | 24033.028 | 7040.671 | 4025.528 | 35936.308 | 16304.188 | 7088.606 |
| set.2.tpm | 39.202902 | 17.963804 | 5.513083 | 239.162028 | 328.822706 | 182.571539 |
| higher.expr | Set 1 | Set 1 | Set 1 | Set 1 | Set 1 | Set 1 |
| log2.ratio | 9.223503 | 8.536321 | 9.271622 | 7.225290 | 5.627408 | 5.271088 |
| precision | 0.9009288 | 0.9094828 | 0.9476440 | 0.5563771 | 0.4609610 | 0.5126263 |
| recall | 0.8712575 | 0.6317365 | 0.5419162 | 0.9011976 | 0.9191617 | 0.6077844 |
| f.score | 0.8858447 | 0.7455830 | 0.6895238 | 0.6880000 | 0.6140000 | 0.5561644 |
- tni-3 is a troponin that is expressed in body wall muscle (BWM) in the head, but not in the posterior.
- cwn-1 and egl-20 are Wnt ligands that are expressed in posterior BWM, but not anterior BWM.
BWM.anterior.vs.posterior.DEG =
two.set.differential.gene.test(
cds,
is.cell.type(cds, "Body wall muscle") &
expresses.gene(cds, "tni-3"),
is.cell.type(cds, "Body wall muscle") &
(expresses.gene(cds, "cwn-1") |
expresses.gene(cds, "egl-20")))
BWM.anterior.vs.posterior.DEG$higher.expr = ifelse(
BWM.anterior.vs.posterior.DEG$higher.expr == "Set 1",
"Anterior BWM", "Posterior BWM")
| gene | set.1.n.umi | set.2.n.umi | set.1.tpm | set.2.tpm | higher.expr | log2.ratio | precision | recall | f.score |
|---|---|---|---|---|---|---|---|---|---|
| tni-13 | 6981 | 93 | 3804.24742 | 18.57887 | Anterior BWM | 7.602169 | 0.9819890 | 1.0000000 | 0.9909127 |
| cwn-1 | 59 | 3113 | 13.99762 | 1311.17230 | Posterior BWM | 6.449980 | 0.9821732 | 0.8968992 | 0.9376013 |
| T21B6.3 | 13869 | 271 | 5966.03306 | 60.20010 | Anterior BWM | 6.607094 | 0.9553265 | 0.8867624 | 0.9197684 |
| glc-4 | 684 | 37 | 310.53057 | 11.4323046 | Anterior BWM | 4.642570 | 0.9506849 | 0.2767145 | 0.4286597 |
| tre-3 | 555 | 5 | 214.79033 | 0.6369876 | Anterior BWM | 7.035742 | 0.9852399 | 0.2129187 | 0.3501639 |
| F48E3.8 | 402 | 1 | 144.96720 | 0.1162907 | Anterior BWM | 7.020870 | 0.9947917 | 0.1523126 | 0.2641770 |
| dpyd-1 | 289 | 6 | 91.93291 | 0.9050051 | Anterior BWM | 5.592715 | 0.9740933 | 0.1499203 | 0.2598480 |
| ceh-34 | 307 | 2 | 131.08432 | 0.2528051 | Anterior BWM | 6.709189 | 0.9885057 | 0.1371611 | 0.2408964 |
| seb-2 | 201 | 5 | 85.08025 | 0.6848107 | Anterior BWM | 5.658166 | 0.9750000 | 0.1244019 | 0.2206506 |
| sfrp-1 | 335 | 2 | 145.18286 | 0.2754131 | Anterior BWM | 6.830763 | 0.9863946 | 0.1156300 | 0.2069950 |
| F35C11.5 | 168 | 1 | 67.97453 | 0.5230727 | Anterior BWM | 5.479937 | 0.9923664 | 0.1036683 | 0.1877256 |
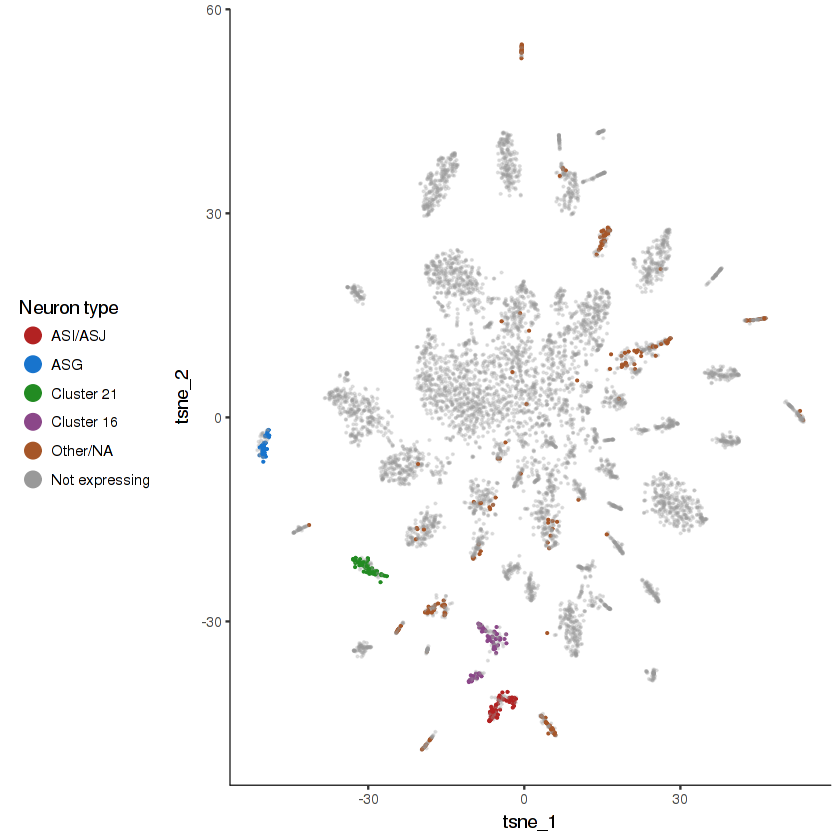
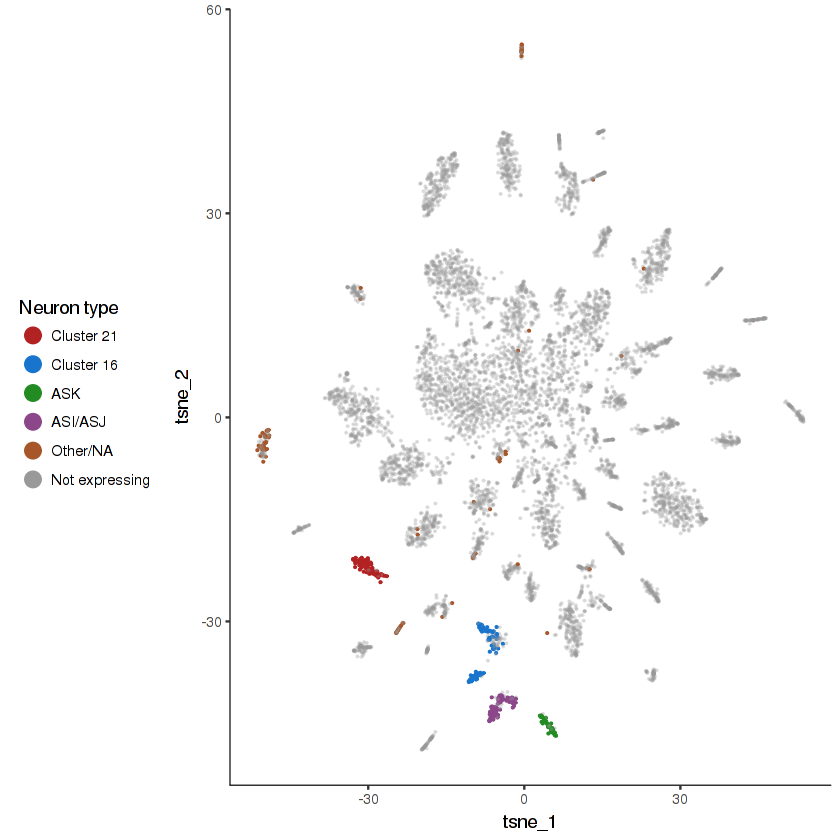
R102.2 is expressed in nine specific pairs of ciliated sensory neurons . We have identified some of these in this dataset, but others remain ambiguous.
show.expr.info("R102.2", "neuron type") %>% head(8)
| facet | tpm | prop.cells.expr | n.umi | total.n.umi.for.facet |
|---|---|---|---|---|
| Cluster 21 | 9807.74014 | 0.91588785 | 446 | 49587 |
| Cluster 16 | 8266.64832 | 0.66025641 | 363 | 47719 |
| ASK | 6720.61429 | 0.81944444 | 155 | 24157 |
| ASI/ASJ | 6482.22237 | 0.74358974 | 239 | 40396 |
| ASG | 2121.04289 | 0.39534884 | 48 | 26295 |
| ASEL | 1670.50501 | 0.37837838 | 17 | 11042 |
| ASER | 795.91129 | 0.28571429 | 16 | 15324 |
| AWB/AWC | 85.09161 | 0.02380952 | 4 | 23186 |
plot.expr(cds.neurons, "R102.2")
two.set.differential.gene.test can be used to find DEG between the mystery R102.2(+) clusters and other ciliated sensory neurons. If you can figure out what these clusters correspond to, let us know!
neuron.cluster.21.vs.other.CSN.DEG =
two.set.differential.gene.test(
cds.neurons,
is.neuron.type(cds.neurons, "Cluster 21"),
!is.neuron.type(cds.neurons, "Cluster 21") &
is.cell.type(cds.neurons, "Ciliated sensory neurons"))
neuron.cluster.21.vs.other.CSN.DEG$higher.expr = ifelse(
neuron.cluster.21.vs.other.CSN.DEG$higher.expr == "Set 1",
"Cluster 21", "Other CSN")
neuron.cluster.21.vs.other.CSN.DEG %>%
filter(higher.expr == "Cluster 21",
precision >= 0.8) %>% head(10)
| gene | set.1.n.umi | set.2.n.umi | set.1.tpm | set.2.tpm | higher.expr | log2.ratio | precision | recall | f.score |
|---|---|---|---|---|---|---|---|---|---|
| C39D10.2 | 226 | 1 | 5237.910 | 1.667334 | Cluster 21 | 10.939377 | 0.9880952 | 0.7757009 | 0.8691099 |
| T09B9.3 | 197 | 0 | 3836.121 | 0.000000 | Cluster 21 | 11.905433 | 1.0000000 | 0.6074766 | 0.7558140 |
| F15A4.5 | 157 | 15 | 3677.812 | 67.963116 | Cluster 21 | 5.736879 | 0.8923077 | 0.5420561 | 0.6744186 |
| flp-25 | 100 | 7 | 1775.299 | 44.020086 | Cluster 21 | 5.301349 | 0.9137931 | 0.4953271 | 0.6424242 |
| C18H7.6 | 118 | 0 | 2385.147 | 0.000000 | Cluster 21 | 11.219863 | 1.0000000 | 0.4579439 | 0.6282051 |
| cdh-3 | 79 | 11 | 1811.618 | 69.489017 | Cluster 21 | 4.683736 | 0.8809524 | 0.3457944 | 0.4966443 |
| K04D7.6 | 65 | 0 | 1110.520 | 0.000000 | Cluster 21 | 10.117019 | 1.0000000 | 0.2710280 | 0.4264706 |
| C29F4.3 | 53 | 0 | 1067.368 | 0.000000 | Cluster 21 | 10.059842 | 1.0000000 | 0.2523364 | 0.4029851 |
| K02E2.1 | 39 | 0 | 1084.714 | 0.000000 | Cluster 21 | 10.083099 | 1.0000000 | 0.2523364 | 0.4029851 |
| dhs-9 | 67 | 15 | 987.323 | 53.024505 | Cluster 21 | 4.191836 | 0.8484848 | 0.2616822 | 0.4000000 |
neuron.cluster.16.vs.other.CSN.DEG =
two.set.differential.gene.test(
cds.neurons,
is.neuron.type(cds.neurons, "Cluster 16"),
!is.neuron.type(cds.neurons, "Cluster 16") &
is.cell.type(cds.neurons,
"Ciliated sensory neurons"))
neuron.cluster.16.vs.other.CSN.DEG$higher.expr = ifelse(
neuron.cluster.16.vs.other.CSN.DEG$higher.expr == "Set 1",
"Cluster 16", "Other CSN")
neuron.cluster.16.vs.other.CSN.DEG %>%
filter(higher.expr == "Cluster 16") %>% head(10)
| gene | set.1.n.umi | set.2.n.umi | set.1.tpm | set.2.tpm | higher.expr | log2.ratio | precision | recall | f.score |
|---|---|---|---|---|---|---|---|---|---|
| F27C1.11 | 223 | 367 | 5302.959 | 1576.21303 | Cluster 16 | 1.7494202 | 0.3537118 | 0.5192308 | 0.4207792 |
| W05F2.7 | 137 | 309 | 3007.794 | 1054.38784 | Cluster 16 | 1.5109326 | 0.3564356 | 0.4615385 | 0.4022346 |
| M04B2.6 | 117 | 4 | 2144.256 | 14.78412 | Cluster 16 | 7.0858594 | 0.9285714 | 0.2500000 | 0.3939394 |
| ocr-2 | 100 | 81 | 2408.536 | 294.50719 | Cluster 16 | 3.0268916 | 0.5465116 | 0.3012821 | 0.3884298 |
| osm-10 | 66 | 32 | 1209.716 | 124.87432 | Cluster 16 | 3.2646129 | 0.7222222 | 0.2500000 | 0.3714286 |
| R102.2 | 363 | 926 | 8266.648 | 3753.48748 | Cluster 16 | 1.1386865 | 0.2524510 | 0.6602564 | 0.3652482 |
| T01D3.1 | 87 | 122 | 1849.955 | 382.64946 | Cluster 16 | 2.2696298 | 0.4112903 | 0.3269231 | 0.3642857 |
| ida-1 | 337 | 1211 | 7843.130 | 5636.27821 | Cluster 16 | 0.4764307 | 0.2234848 | 0.7564103 | 0.3450292 |
| lap-2 | 132 | 55 | 2344.214 | 231.93208 | Cluster 16 | 3.3311228 | 0.5263158 | 0.2564103 | 0.3448276 |
| rps-11 | 83 | 230 | 2013.967 | 1036.83972 | Cluster 16 | 0.9564565 | 0.2863636 | 0.4038462 | 0.3351064 |
Use Case 4: Re-clustering cell subsets using t-SNE
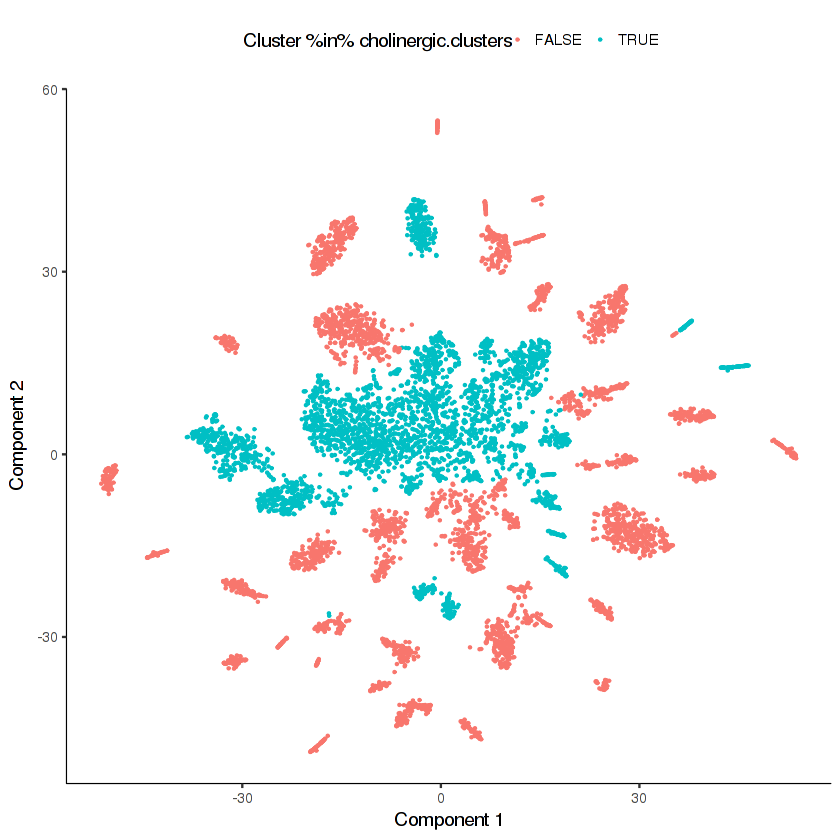
In the Cao et al. paper, we found that several of the neuron t-SNE clusters expressed markers of cholinergic neurons such as unc-17, cho-1, and cha-1. Let's perform a sub-clustering of these cholinergic neurons to see if we can get better separation. First, we'll identify which clusters are enriched for cells that express cholinergic markers.
pData(cds.neurons)$any.cholinergic.marker =
(expresses.gene(cds.neurons, "unc-17") +
expresses.gene(cds.neurons, "cho-1") +
expresses.gene(cds.neurons, "cha-1")) > 0
pData(cds.neurons) %>%
group_by(Cluster) %>%
summarize(
n.total = n(), n.cholinergic =
sum(any.cholinergic.marker),
prop.cholinergic = n.cholinergic / n.total) %>%
inner_join(
unique(pData(cds.neurons)[, c("Cluster",
"neuron.type")]),
by = "Cluster") %>%
arrange(-prop.cholinergic) %>%
head(15)
| Cluster | n.total | n.cholinergic | prop.cholinergic | neuron.type |
|---|---|---|---|---|
| 29 | 305 | 155 | 0.5081967 | Cholinergic (29) |
| 23 | 45 | 19 | 0.4222222 | Cholinergic (23) |
| 3 | 385 | 131 | 0.3402597 | Cholinergic (3) |
| 26 | 261 | 81 | 0.3103448 | Cholinergic (26) |
| 35 | 58 | 16 | 0.2758621 | Cholinergic (35) |
| 36 | 128 | 35 | 0.2734375 | Cholinergic (36) |
| 15 | 65 | 16 | 0.2461538 | Cholinergic (15) |
| 12 | 68 | 14 | 0.2058824 | DVA |
| 24 | 188 | 38 | 0.2021277 | Cholinergic (24) |
| 8 | 117 | 23 | 0.1965812 | ASI/ASJ |
| 11 | 1998 | 387 | 0.1936937 | Cholinergic (11) |
| 6 | 160 | 21 | 0.1312500 | SDQ/ALN/PLN |
| 25 | 363 | 43 | 0.1184573 | Cluster 25 |
| 41 | 211 | 24 | 0.1137441 | Doublets |
| 16 | 156 | 17 | 0.1089744 | Cluster 16 |
cholinergic.clusters = c(29, 23, 3, 26, 35, 36, 15, 24, 11)
plot_cell_clusters(cds.neurons, color =
"Cluster %in% cholinergic.clusters",
cell_size = 0.2)
cds.cholinergic =
cds.neurons[, pData(cds.neurons)$Cluster %in%
cholinergic.clusters]
cat(ncol(cds.cholinergic), "cells in the cds subset", "\n")
cds.cholinergic = estimateSizeFactors(cds.cholinergic)
cds.cholinergic = estimateDispersions(cds.cholinergic)
# above line would take a lot of memory for larger cell sets
cds.cholinergic = detectGenes(cds.cholinergic, 0.1)
> 3433 cells in the cds subset
The next step is to run a new t-SNE dimensionality reduction on this subset of cells.
system.time({
cds.cholinergic = reduceDimension(
cds.cholinergic, max_components = 2, norm_method = "log",
num_dim = 20, reduction_method = 'tSNE', verbose = T)
})
pData(cds.cholinergic)$tsne_1 = reducedDimA(cds.cholinergic)[1,]
pData(cds.cholinergic)$tsne_2 = reducedDimA(cds.cholinergic)[2,]
| user | system | elapsed |
|---|---|---|
| 164.398 | 2.898 | 168.483 |
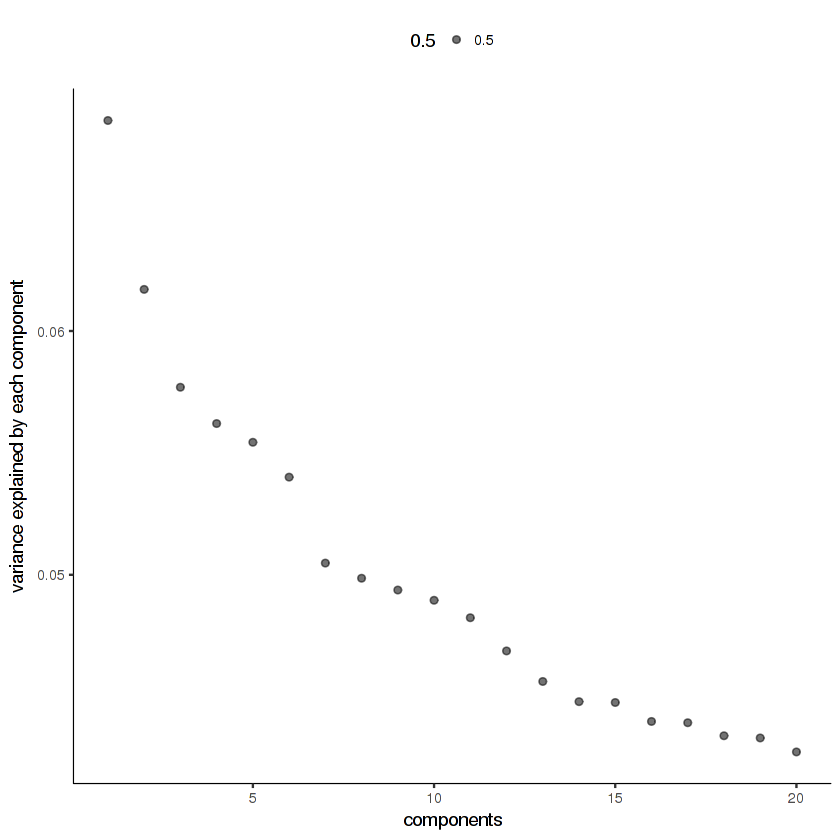
20 principal components looks like it's enough for this data. If you don't see an elbow in the scree plot, that means you've used too few principal components.
plot_pc_variance_explained(cds.cholinergic)
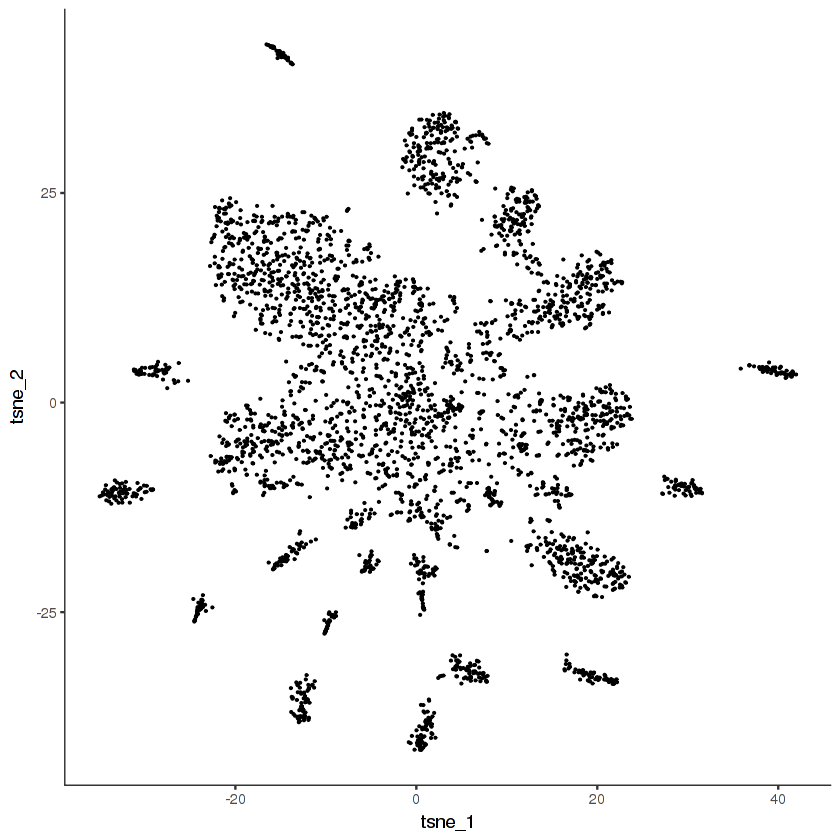
ggplot(pData(cds.cholinergic), aes(x = tsne_1, y = tsne_2)) +
geom_point(size = 0.1) +
monocle:::monocle_theme_opts()
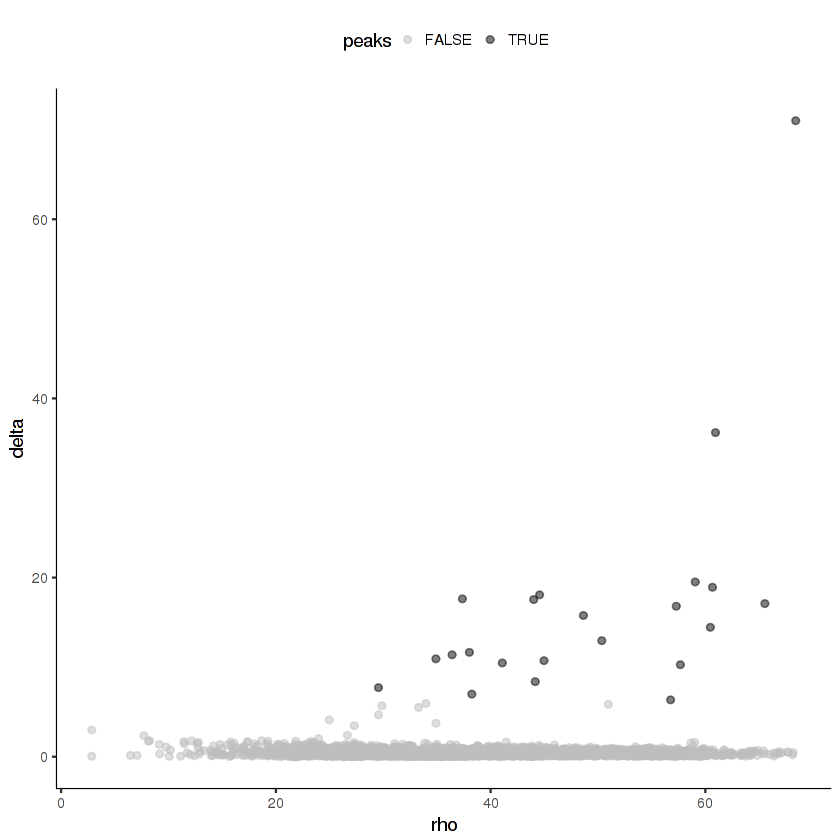
Now we'll cluster the cells in the t-SNE space using density peak clustering. In density peak clustering, rho measures to the local density of cells and delta measures the minimum distance to a region of higher local density. Go to this site for more details. You want to set thresholds on rho and delta that enclose the outlier points in the scatter plot. In my experience, it's usually better to over-cluster than to under-cluster.
cds.cholinergic = clusterCells_Density_Peak(cds.cholinergic)
> Distance cutoff calculated to 2.694535
plot_rho_delta(cds.cholinergic,
rho_threshold = 10,
delta_threshold = 6)
cds.cholinergic = clusterCells_Density_Peak(cds.cholinergic,
rho_threshold = 10, delta_threshold = 6, skip_rho_sigma = T)
plot_cell_clusters(cds.cholinergic, cell_size = 0.2)
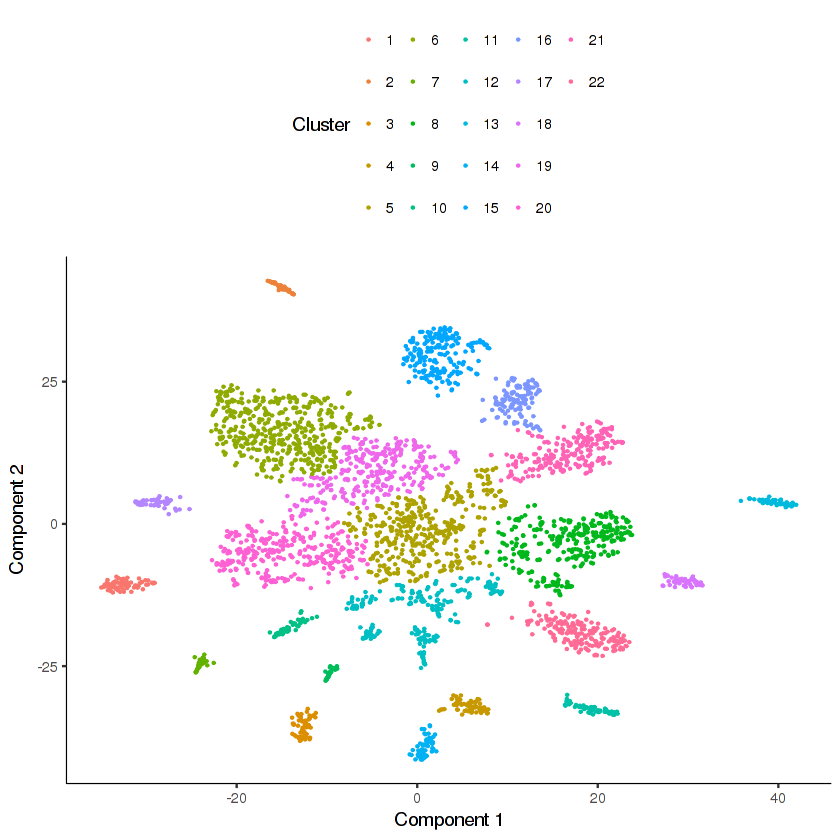
Looks like there are many distinct clusters. Recall that we input only 9 clusters from the cds.neurons t-SNE into this re-clustering. The re-clustering appears to have revealed new potential distinct cell types.
Now would be a good time to save your progress.
save.image("my_analysis_Cao_et_al_data.RData")
Let's find marker genes for each cluster.
cholinergic.clusters =
sort(as.integer(unique(pData(cds.cholinergic)$Cluster)))
cholinergic.clusters
> 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
DEG.results = lapply(cholinergic.clusters,
function(this.cluster) {
message("Finding markers for cluster ", this.cluster)
cbind(
two.set.differential.gene.test(
cds.cholinergic,
pData(cds.cholinergic)$Cluster == this.cluster,
pData(cds.cholinergic)$Cluster != this.cluster),
data.frame(cluster = this.cluster))
})
cholinergic.cluster.markers =
do.call(rbind, DEG.results) %>%
filter(higher.expr == "Set 1") %>%
select(
cluster, gene,
cluster.n.umi = set.1.n.umi,
other.n.umi = set.2.n.umi,
cluster.tpm = set.1.tpm,
other.tpm = set.2.tpm,
log2.ratio, precision, recall, f.score) %>%
arrange(-f.score)
cholinergic.cluster.markers %>%
filter(precision >= 0.5) %>% head(10)
| Cluster | gene | cluster.n.umi | cluster.n.omi | cluster.tpm | other.tpm | log2.ratio | precision | recall | f.score |
|---|---|---|---|---|---|---|---|---|---|
| 7 | B0432.14 | 194 | 11 | 10886.411 | 5.8332893 | 10.637661 | 0.9047619 | 0.9500000 | 0.9268293 |
| 13 | nlp-42 | 299 | 15 | 22725.799 | 17.1886827 | 10.287074 | 0.8833333 | 0.9137931 | 0.8983051 |
| 13 | nlp-42 | 299 | 15 | 22725.799 | 17.1886827 | 10.287074 | 0.8833333 | 0.9137931 | 0.8983051 |
| 21 | flp-12 | 1379 | 297 | 17963.487 | 366.8397815 | 5.609846 | 0.7282609 | 0.8777293 | 0.7960396 |
| 13 | T04C12.3 | 173 | 25 | 12067.242 | 22.8874212 | 8.980629 | 0.7407407 | 0.6896552 | 0.7142857 |
| 22 | lgc-39 | 289 | 93 | 5300.848 | 120.7416859 | 5.444328 | 0.6346154 | 0.6839378 | 0.6583541 |
| 1 | sem-2 | 128 | 63 | 6240.354 | 52.4115889 | 6.868331 | 0.6615385 | 0.6056338 | 0.6323529 |
| 2 | vglu-2 | 30 | 1 | 2356.216 | 0.6977763 | 10.438609 | 0.9523810 | 0.4444444 | 0.6060606 |
| 15 | Y48C3A.5 | 339 | 145 | 6989.459 | 148.0684921 | 5.551134 | 0.5747664 | 0.6243655 | 0.5985401 |
| 10 | glb-17 | 81 | 19 | 4790.632 | 16.9955396 | 8.056433 | 0.7352941 | 0.5000000 | 0.5952381 |
| 9 | nlp-5 | 34 | 24 | 3552.050 | 21.1320088 | 7.326374 | 0.5500000 | 0.6470588 | 0.5945946 |
In the previous examples for using plot.expr, the function used pre-computed expression tables for cds and cds.neurons to show which cell types / neuron types were expressing a given gene. The following code will set up plot.expr so it can show which density peak clusters from this re-clustering analysis express a given gene. You can also use show.expr.info with your own clustering.
cholinergic.expr.info = get.expr.info.by.facet(cds.cholinergic,
"Cluster")
plot.expr(cds.cholinergic, "lgc-39", expr.info = cholinergic.expr.info)
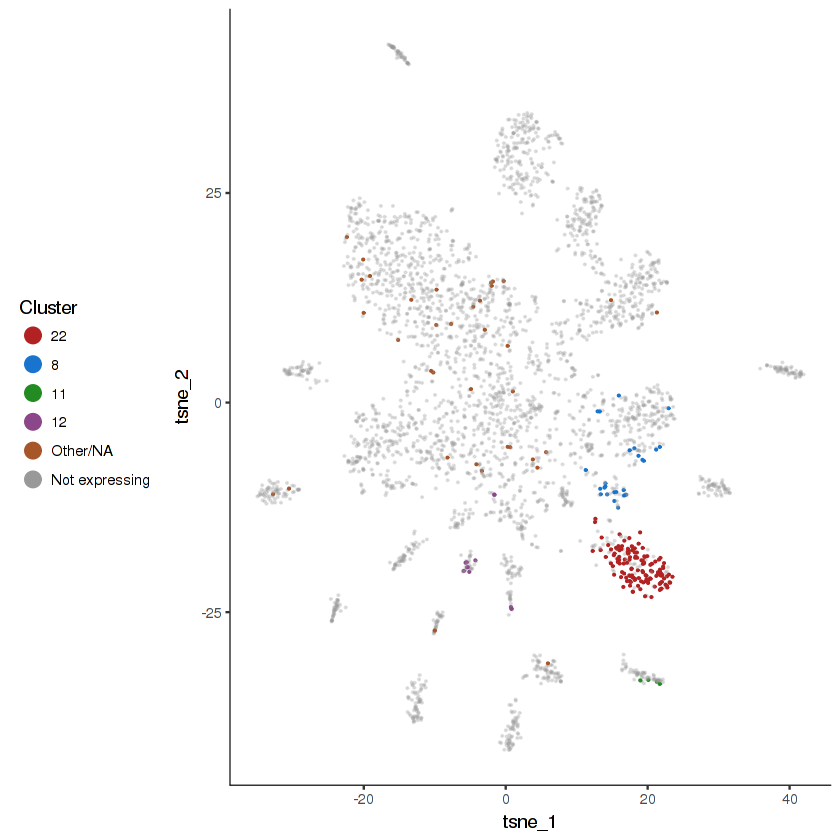
show.expr.info("lgc-39", cholinergic.expr.info) %>% head(10)
| facet | tpm | prop.cells.expr | n.umi | total.n.umi.for.facet |
|---|---|---|---|---|
| 21 | 4677.68319 | 0.618644068 | 305 | 63153 |
| 18 | 244.90291 | 0.060606061 | 4 | 14652 |
| 19 | 232.84907 | 0.052884615 | 14 | 50800 |
| 8 | 217.45530 | 0.038461538 | 18 | 80071 |
| 4 | 155.65487 | 0.027100271 | 24 | 174660 |
| 22 | 129.10174 | 0.024615385 | 10 | 117663 |
| 1 | 79.17593 | 0.028169014 | 2 | 19954 |
| 6 | 60.42661 | 0.014925373 | 1 | 17215 |
| 9 | 56.34438 | 0.029411765 | 1 | 12093 |
| 10 | 30.47108 | 0.008196721 | 1 | 26202 |
plot.expr(cds.cholinergic, "vglu-2", expr.info = cholinergic.expr.info)
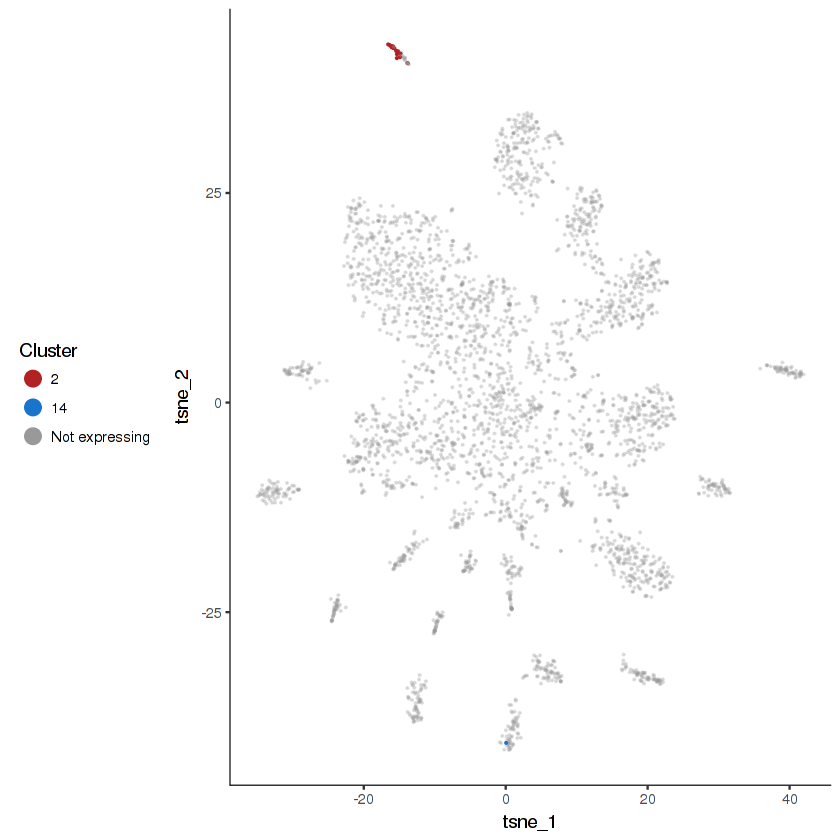
show.expr.info("vglu-2", cholinergic.expr.info) %>% head(10)
| facet | tpm | prop.cells.expr | n.umi | total.n.umi.for.facet |
|---|---|---|---|---|
| 2 | 2356.21566 | 0.44444444 | 30 | 11540 |
| 14 | 37.52486 | 0.01587302 | 1 | 23493 |
| 1 | 0.00000 | 0.00000000 | 0 | 19954 |
| 3 | 0.00000 | 0.00000000 | 0 | 19911 |
| 4 | 0.00000 | 0.00000000 | 0 | 174660 |
| 5 | 0.00000 | 0.00000000 | 0 | 51233 |
| 6 | 0.00000 | 0.00000000 | 0 | 17215 |
| 7 | 0.00000 | 0.00000000 | 0 | 17691 |
| 8 | 0.00000 | 0.00000000 | 0 | 80071 |
| 9 | 0.00000 | 0.00000000 | 0 | 12093 |
plot.expr(cds.cholinergic, "unc-17", expr.info = cholinergic.expr.info)
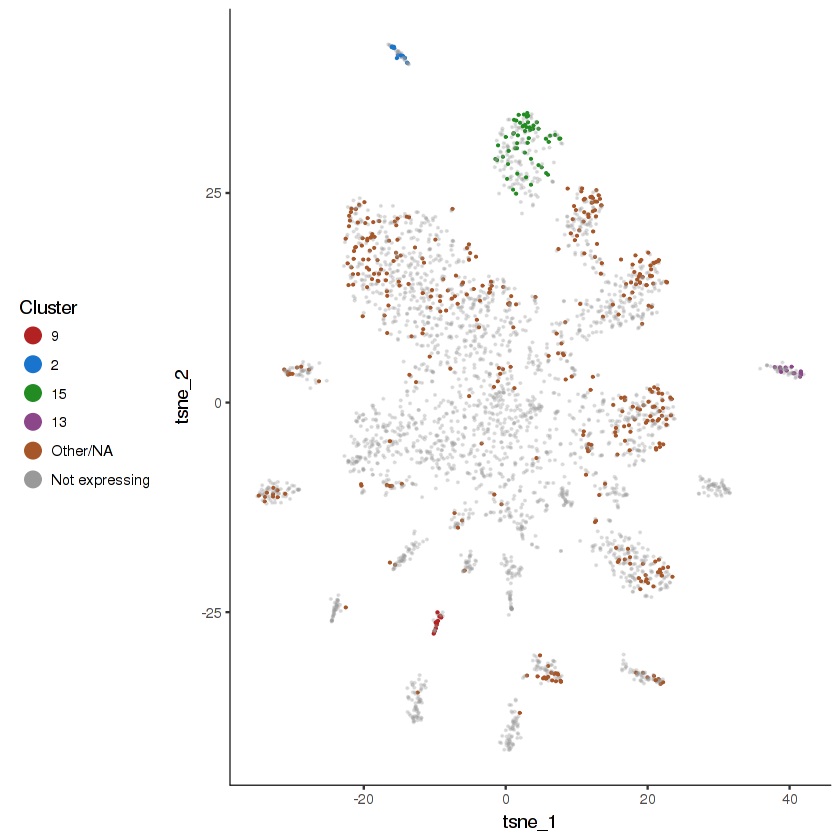
show.expr.info("unc-17", cholinergic.expr.info) %>% head(10)
| facet | tpm | prop.cells.expr | n.umi | total.n.umi.for.facet |
|---|---|---|---|---|
| 9 | 3200.3642 | 0.3823529 | 44 | 12093 |
| 2 | 2944.3520 | 0.2888889 | 27 | 11540 |
| 13 | 2128.1049 | 0.2500000 | 29 | 13140 |
| 5 | 2087.7739 | 0.2731959 | 119 | 51233 |
| 15 | 1992.1189 | 0.2941176 | 62 | 31426 |
| 6 | 1721.6386 | 0.2686567 | 34 | 17215 |
| 8 | 1527.0847 | 0.1826923 | 126 | 80071 |
| 4 | 1344.6852 | 0.1585366 | 235 | 174660 |
| 12 | 1114.1395 | 0.1718213 | 123 | 97104 |
| 16 | 886.1635 | 0.1923077 | 16 | 19103 |